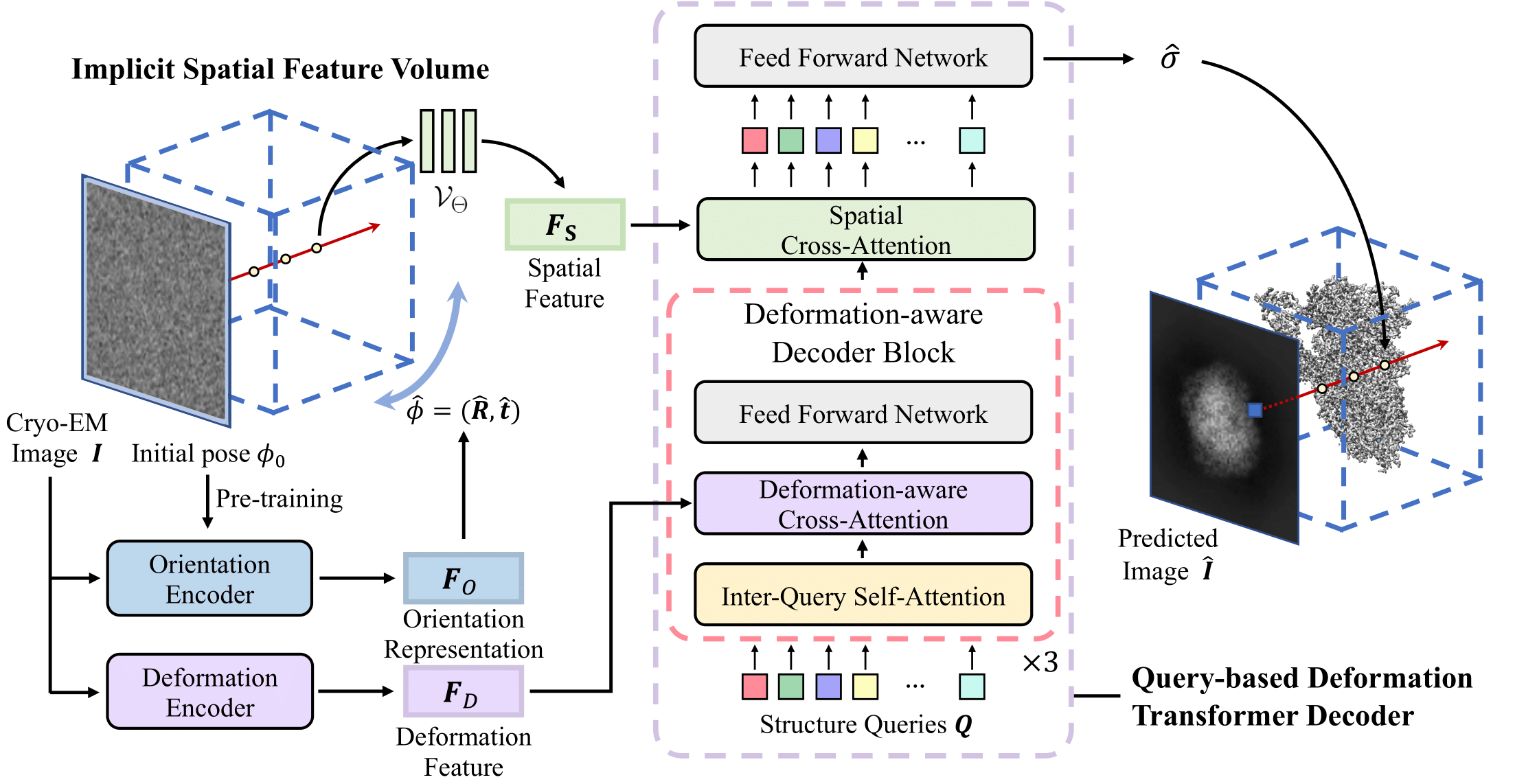
Abstract
Cryo-electron microscopy (cryo-EM) allows for the high-resolution reconstruction of 3D structures of proteins and other biomolecules. Successful reconstruction of both shape and movement greatly helps understand the fundamental processes of life. However, it is still challenging to reconstruct the continuous motions of 3D structures from hundreds of thousands of noisy and randomly oriented 2D cryo-EM images. Recent advancements use Fourier domain coordinate-based neural networks to continuously model 3D conformations, yet they often struggle to capture local flexible regions accurately. We propose CryoFormer, a new approach for continuous heterogeneous cryo-EM reconstruction. Our approach leverages an implicit feature volume directly in the real domain as the 3D representation. We further introduce a novel query-based deformation transformer decoder to improve the reconstruction quality. Our approach is capable of refining pre-computed pose estimations and locating flexible regions. In experiments, our method outperforms current approaches on three public datasets (1 synthetic and 2 experimental) and a new synthetic dataset of PEDV spike protein.